There are plenty of tools for development, but I really wanted to take advantage of LXD for web development, so I ended up writing ll4d: the LXD(LXC)-LAMP environment For Development environment.
There are probably better ways to do this, and highly optimized juju charms to build something much better. But for my persona use case, this script is just perfect. It takes just 48 seconds to build everything and it has 100% all the functionality I need.
It is a very simple script, which works as follows:
- Sets up a basic LXC/LXD environment
- Creates a container with a LAMP (Linux Apache MySQL PHP) server
- Creates a fancy symbolic link from the apache root directory at the LAMP container to somewhere in your home or desktop. This allows you to work on your code using your visual tools and view the result on the fly
- Create a simple website to test Apache, PHP and MySQL
Here is an screenshot of a working environment. You work from a directory directly under your home, access the LAMP server for working with configurations and MySQL databases and access the server on at the given IP.
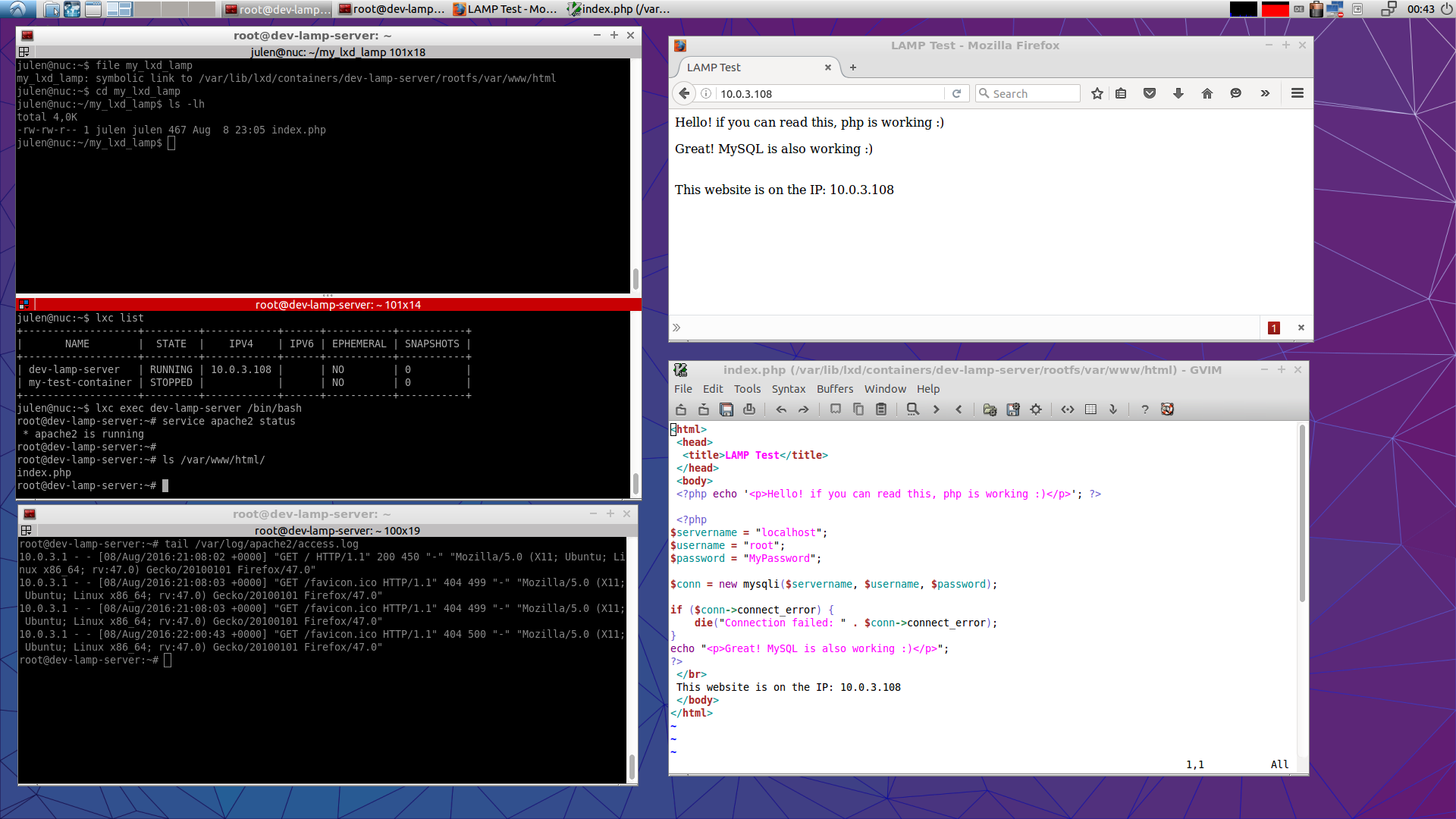
Screenshot of a work environment with ll4d.
ll4d is available on my github repository: https://github.com/julenl/ll4d
The few instructions on how to download and exectute it, can be found at the README file.
I hope you find it useful… if you don’t, you can remove it completely in 3 simple steps:
./ll4d.sh –clean
rm -rf ll4d*
sudo aptitude purge lxd