Rise your hand if you have ever gotten a pseudo-potential from somewhere and you started to use it just by checking the recommended values of the cutoffs inside the file.
Are we sure these values are good?
If you are an skeptic scientist and you do not thrust what you don’t do by yourself, now there is a really simple and cheap way for you to test for the optimum cutoff of any pseudopotential with Quantum Espresso: my pw_cutoff checking script.
The ecutwfc is described in the manual as the kinetic energy cutoff for wave functions in Ry.
So, when we make a Density Functional calculation with plane waves, the (electronic) wavefunction is represented as the infinite summation of plane waves, which must be truncated, in order to be able to handle them computationaly.
So, the more plane waves, the more accuracy, but also higher computational cost. The trick for speeding up the calculations relies on the fact that the plane waves with less kinetic energy (represented as ℏ2 / 2 m |k+G|2) have higher contribution to the totality, so the plane waves with lower energy are the most important. But, up to which point should we increase the cutoff, so that we get a good balance between computational cost and accuracy? That is precisely the optimum cutoff choice.
In the practice, this choice can be done just by calculating a single point (wavefunction optimization) at a range of energy cutoffs, and the best choice will be the one which shows really small changes in the total energy with respect to the cutoff.
This task is now automated with the pw_cutoff script.
Let’s make a quick check on the optimum cutoff for some pseudopotential.
It runs simply as:
./pw_cutoff.sh -f ~/pwpsp/H.pz-rrkjus.UPF
where -f is the option for specifying the pseudo potential we want to test. For more options check the -h option.
From the following snapshot, it can be appreciated that the test runs quite fast (about 2 minutes) on a desktop computer (Intel i7/Ubuntu):
Snapshot of the usage of the pw_cutoff script.
The script will try to find out if we are using ultra-soft pseudo potentials and use an special set of cutoff values in that case (lower ones). It will also pop up, and save a graph with the representation of the results looking like this one:
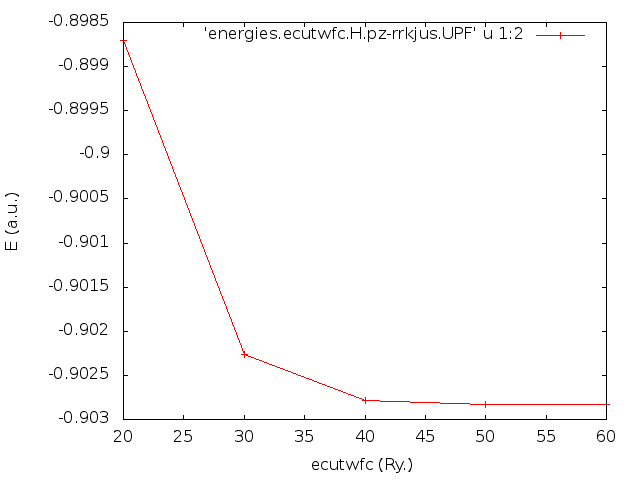
Output figure from pw_cutoff script.
In this case, the script detected that the pseudo potential was ultra soft, and defined a set of cutoffs from 20 to 60 Ry.
As we can appreciate in the figure, the best choice for the ecutwfc value seems to be 40 Ry for this pseudo potential.
You should run this script for all the different pseudo potentials you are using in your polyatomic system, and set the ecutwfc to the highest value obtained.
In the next graph you can see the impact of the cutoff in the calculation time.

ecutwfc value vs calculation time graph.
The time needed to complete the calculation in this case is actually bigger for the lower cutoffs. This is because the basis is so bad that the self consistent calculation needs more cycles in order to converge to the final result. But as it can be seen, the time increases after 40 Ry, so it is not worth to pay such a computational cost for such a tiny improvement in the result.
On the next “episode” of pw_cutoff I will probably write something to find the optimum cutoff for the charges (ecutrho) as well 🙂
Hello Sir
I am muthu. currently working as project fellow in Dept. of Theore. Phys. Madurai kamaraj University,India. really i am impressed about your website and scripts.
i’m using quantum espresso for my project work. slowly i am learning this. i have one request. i want to create slab and wire of tio2 (this is aim of my current project work). i do not have well sophisticated system or software.( i do not like to purchase costly items like material studio, frankly i can not buy).
so i request you to give your valuable suggestion on how to create slab and wire using Avogadro or vesta ( these are the softwares i have in my pc).?
which software is highly reliable?
what are the other software to do the above work?
currently i focus on this so your opinion will be highly helpful.
i eagerly wait for your hearing and reply
thank you
Dear Muthu,
When working with crystal or symmetric structures I usually build my systems by writing small scripts to repeat the atoms in x, y and z.
Crystal structures are faster to build without Graphical User Interface.
So, my suggestion is to invest some time in learning some scripting language (python, bash, perl, …), because no visualization or analysis program is as effective as a good self made script. Also, a molecular modeler which can not write scripts or programs, is like a carpenter which can not cut wood.
If you still want to use a GUI, Avogadro or XCrysten are a good choice. The good thing of XCrysden is, that it offers a good integration with Quantum Espresso inputs and outputs.
You can download the TiO2 crystal structure from some open crystal structure database and modify it with XCrysden in order to create your initial structure.
You don’t really need to buy any software package. Most (if not all) of the software we use is free (open source), or free of charge for academics.
If you ask me for some software for calculating TiO2, so DFT based, I would personally suggest Quantum Espresso (open source) or CPMD (free of charge for academics).
Best regards
Loved it!
helped me too! I searched a lot for this tool, it was in hand. Besides, your shell script has some structures unknown to me and be like a lesson script too.
Congratulations on the website and the valuable information and scripts.
Alex.
Theoretical chemistry studant.
Brazil.
Thank you for the script! it’s really useful.
Dear larrucea
I use this script but I show this error.
ERROR: Provide a path to a pw.x binary.
How I use from this script?
your sripts is really useful
Thanks so much
Elham
Theoretical chemistry student
Iran
Dear Elham,
This script just generates a basic input file for Quantum Espresso with your chosen pseudopotential, then runs some quick calculations with that input using different values for the cutoff, and finally shows you a graph with the results. But for running those calculations, you need to have a Quantum Espresso binary in your system, and the path to that binary should be given after the “-x” flag.
Did you check the “-h” (help) option?
Good luck!
Julen
Dear larrucea
I want to create a column of three discotic molecule but i cant specify lattice parameters in &SYSTEM.
when i try to visualize my input file in XCrysden, i am faced with a irregular and disarrenged pattern in the unit cell!
How can I deal with rather big molecules in Quantum Espresso? how to define the correct unit cell and supercells?
i eagerly wait for your response
thank you
Dear Pirvaz,
Number 1: check the literature and the crystal databases.
If you can’t find it there, proceed to try to build it yourself.
If you are dealing with crystal structure you could simply do some math on a piece of paper.
You can make an approximate guess of the height if each molecule (perhaps ~2 Å up and down?), and build a test structure. You can do that by:
awk '{print $1 $2 $3 $4+1}' your_str.xyz > new_str.xyz… and append the content of file new_str.xyz to your xyz file (and increase the number of atoms at the first line).
Calculate a single point of that structure together with it’s stress tensor, and put the layers closer or further depending on whether you want it more or less packed.
Good luck
As a beginner this information about energy cut-off is more valuable. Thanks a lot.
Dear Julen
I just happened to visit this site while seeking answers to my questions. I have tried your suggestions to test the pseudopotentials and it has worked prefectly fine. Thank you for this wonderful idea!!!
I however want to understand the following.
1) My system is having 232 atoms (including 4 Co atoms H’s,C’s,N’s in a 1D chain) and normally if I have to calculate the properties like spin-den I have to fix the parameters first and do the convergence tests. Before I came across this site I had done single point calculations on my system for 2 cases: Ecutwfc = 35.0 and Ecutrho = 270.0 && Ecutwfc = 40.0 and Ecutrho = 320.0. I used the pseudo potentials .pbe-rrkjus.UPF for all the 4 types of atoms. It takes about ~5-6 days to complete on a 32c job. I got optimum values as H,Co = 40 Ry & C,N = 30 Ry. The job with Ecutwfc = 40 converged faster.
Q: Is what you have described above the same as running convergence tests to fix cut values? …for a given set of pseudopotentials.
If it be so then I can save many days of test runs on my job. PLease answer my question asap.
Thanks
Michael
jncasr
Hi Michael,
Sorry for the late reply. Yes, that’s exactly what it does, it calculates single points at different cutoffs and plots a graph, so that you can estimate which is the optimum value.
I am glad I could help 🙂
Sir , I am learning the quantum espresso but i have some obstacles to understand the ecutwfc for a molecule. I want to ask how to choose the kinetic energy cut off for a molecule because molecule has different type of atoms and I think ecutwfc depend on atoms.My problem is that ,can we take ecutwfc same for all atom.
Hi Gurav,
You should find the optimum cutoff for all different species (atoms types). The higher the value the more accurate the calculation, but the higher the cost…
Dear Sir
First of all thank you for the valuable information you include in your site. I believe this helps a lot of us trying to get a grasp of QE and quantum chemical methods in general.
My question is a general cuestión on optimización of input parameters. First of all I somehow thought that cutoff energy optimisation should be done with the whole system and not only whith the PP. Does the crystal structure affect on the parameter optimisation or its impact is so low that it doesn’t affect much?
Another more general question is: should there be an order for optimisation operations? i.e. First relax the cell, then optimise cutoff, then relax the atom positions? Or some other order?
Thank you in advance for your answer
Hi Ion,
The pseudopotentials describe each specie independently, so you need to set a cutoff for each of them separately. I think you might be confusing the cutoff for the pseudopotentials and the cutoff for the energy optimization. This second one describes the difference in the total energy compared to the previous SCF or geometry optimization step. This means that when the difference of energy from the current step compared to the previous one is “very small”, you can assume the wavefunction or geometry to be converged.
If I think of a very rough and inaccurate analogy, think of the pseudopotential cutoff as the “amount of accuracy” to describe the atom. If you are working on crystals, you need the atoms to be very accurately described, so it’s good to use a little higher cutoffs.
About the second question. You can do it in any order, but I think you are a little confused. You don’t need to optimize the cutoff of the pseudopotentials, you do that once, or just use whatever you find in the literature or suggested in the pseudopotential file.
I know of 3 optimization processes:
– Wavefunction: calculate the wavefunction coefficients > change them a little bit and calculate a again > if better than previos result, keep changing in this direction, if not try in some other direction > when the difference between the total energy compared to the previous one is “small” (Energy cutoff), the wavefunction “is converged”
– Geometry: Make the previous step to calculate the wavefunction > calculate the forces and move the atoms to places where they are more relaxed > calculate wavefunction again > if the energy difference with previous step is “small” (geometry cutoff) the structure is converged (optimized)
– Cell: optimize geometry > try to change the lattice parameters to make the total energy of the cell be as low as possible > iterate until converged
You can do a cell optimization by hand (since they are very expensive computationally), by optimizing the geometry of your crystal, changing the cell parameters up and down, and see which one is the lowest.
Being a fledgling person in Quantum espresso, I am finding it difficult to how to incorporate your code for Cut off calculation. Is the shell script required to be generated to use your code or how else can i use it. Kindly help me with the steps to use it.
Hi Chayan
Nope. The script just generates input files and runs them with the pw.x binary provided by you. So, you basically give the pseudo-potential and it returns the values for you to chose which is the “converged enough” value.
Dear Julen.
Thank you so much for this script. It’s very usefull. I have nontrivial question to you 🙂 I’m working on some paper, and I want to write something like that: “optimal cut off energy was obtined by pw_cutoff program [238]”.
How can I cite yours program?
Yours sincerely,
Miłosz Martynow
Hi Miłosz
Thank you very much! I guess you could put a link to the website 🙂
Excuse me, i’m trying to use your pw_cutoff program, but I can’t because it said: No such file or directory.
I put your program and the pseudopotential file in the same folder, so first I typed this command:
./pw_cutoff.sh -f Ba.pbe-spn-kjpaw_psl.1.0.0.UPF
It didn’t work, then I wrote the whole pathname of the folder like this:
./pw_cutoff.sh -f /home/son/BaTiO3/Ba.pbe-spn-kjpaw_psl.1.0.0.UPF
Still didn’t work, could you help me please?
The “pw.x” (quantum espresso) binary, has to be on your path.
I still have 2 questions here,
First, the pw_cutoff script is still in /home/BaTiO3. Do I have to move your pw_cutoff script into /bin folder?
Second, if the path of my /bin folder is /home/qe-6.2.1/bin, the path of pseudopotential file is /home/BaTiO3/Ba.pbe-spn-kjpaw_psl.1.0.0.UPF (QE and BaTiO3 are different folders in /home), what should I type in the pw_cutoff command?
Thank you very much.
It’s been really long since I used to work with stuff, but if I remember right, there should be some environment variable or something you can set as the path to your pseudopotentials file. You don’t have to copy the pw_cutoff to the /bin, just execute it as “./pw_cutoff”, like any other script. If you put it in /bin you can save the “./”, but it’s not necessary.
Sorry for my late reply, but i’ve done it, with your instruction. Thank you very much. Can’t wait for your ecutrho script! 😀
Dear Son Nguyen
please, can you till me how did you solve that, I have the same Error here
Hi Dr Julen
I want to use your script to find ecutwfc, everything was ok until the end, it said that:
…
calculating ecutwfc=110 …
calculating ecutwfc=120 …
./pw_cutoff.sh: line 153: d: command not found
– DONE! –
Then i looked into your script, around line 153:
…
else
d $ECHO “No gnuplot in PATH. Results not plotted.”
fi
else
…
you can see that in line 153, there is a letter “d” in front of $ECHO. I don’t know what kind of program language that you wrote so i don’t know what does letter “d” mean, but are there any problem in line 153? Please let me know as soon as possiblel, because i’m doing my thesis.
Thank you very much
That “d” is not supposed to be there. Someone did a mistake copy/pasting…
Hi Julen,
Thank you very Much. From University of Ghana, Materials Sciences Department.
NAD
Thank you so much for your excellent work. Could you provide for another scripts for K-points and lattice constants convergence test.