Infrared (IR) and Raman spectra are two very valuable tools for the characterization of chemical compounds. And although there seem to be many different possibilities to produce them computationally, I didn’t find any clear tutorial on how to produce them using free software tools. Some of them, give complicated instructions on how to calculate second derivatives or Raman tensors, or stuff like that, but in the end one wonders: Where are the numbers I have to plot!
…Thereby, here is my post about computing IR and Raman spectra with Quantum Espresso.
For the impatient: most of this post is resumed in my Quantum Espresso example for the PHonon code, which will run for some 15-17 minutes and pop-up the IR/Raman spectra of CO2 and ZnO (Wurtzite).
If you are familiar with Quantum espresso, this will be very simple for you.
The procedure is as follows:
- Optimize the wavefunction by performing an Self Consistent Field (scf) calculation with pw.x
- Calculate the vibrational frequencies (normal modes/phonons) with ph.x
- Extract the phonon information from ph.x output using dynmat.x
- Parse the dynmat.x output section that contains the spectra data (frequencies and intensities) and plot it with gnuplot, producing these two spectra:
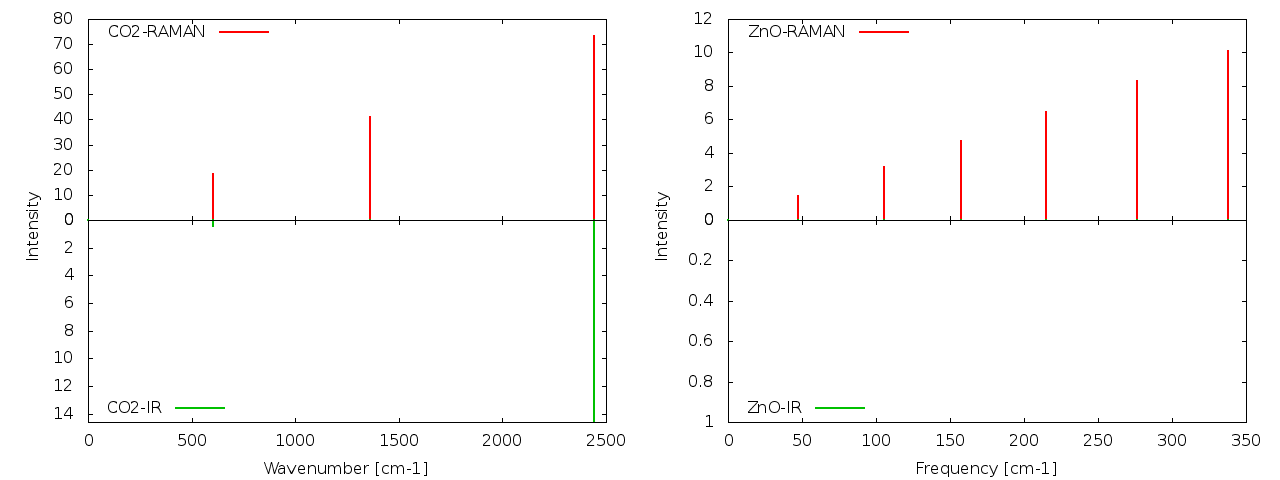
IR and Raman spectra for Co2 and ZnO, produced by Quantum Espresso tools.
The obtained results for CO2 show normal modes at 604.82 (bending), 1362.90 (symmetric stretching) and 2441.15 cm-1 (antisymmetric stretching) which agree very well with the expected values of 667, 1340 and 2349 cm-1.
For the case of ZnO, the results can be compared to the ones by Calzolari & Nardelli and despite this calculation is a really rough approach intended to get some quick and dirty result, it already shows some similarities in the tendency of the lower frequencies.
There are some vibrational modes missing. This occurs because we only used 4 atoms to simulate an infinite bulk structure, and that creates some error. In the article I mentioned above, they use 512 atoms and besides, higher cutoffs and larger amount of k-points. But that calculation can not be run on a regular PC 😉
Thank you so much! This was extremely helpful. Regards, Keith Prisbrey, Prof. Emeritus, Materials Science and Engineering, University of Idaho, USA
Dear Dr. Larrucea,
I have followed the example015 in the PHonon directory regarding the IR and Raman spectra of CO2 and ZnO. First off, I have to thank you for your explanations to this example. However, I have realized that in the CO2.dm.out file there are some frequencies having negative values (which so far I have realized there is no physical meaning for that ). My questions are:
1. Does it suffice to extract those numbers in the dm.out files and plot them as IR and Raman spectra?
2. What should we do in order not to get negative frequencies?
I want to do the exact calculations for SOD (a kinda of zeolite). Is there another way to get the spectrum or I can follow the same procedure?
I would really appreciate it if I could use your help and experience for my questions.
Best regards,
Amir M. Mofrad
Graduate Research Assistant
Chemical Engineering Department
University of Missouri
Dear Amir,
Sorry for the delay.
It’s been a while since I haven’t been dealing with these things, but well… in principle those frequencies in the dm.out file should be the ones to use.
The negative frequencies are usually because the structure is not right. However, sometimes, if you have a relatively large amount of atoms and you get one or two negative frequencies, they could be neglected. But for the CO2, you better try to converge the geometry a little better, by increasing cutoffs.
Also, I noticed that I did a mistake regarding the intensities when plotting the spectra. So the frequencies (x axis) should be OK, but the intensities (y axis) are not.
Dear Dr. Larrucea,
Thank you very much for your detailed example. It helps me a lot. But how can I get the intensities of Raman spectra.
Hi Chunyi,
It’s been really long since I did this things. Better as in the quantum espresso mailing list, they will give you a better answer.
Hi. I am wondering how I can know the vibrational mode from the output. For example, in Gaussian, wen can check the vibrational mode by Gaussview.
Hi Yuanquing,
You can also visualize those modes using some visualization program, like jmol or VMD.
Dear Julen,
Does Quantum espresso can print dipole moment derivatives and polarizability derivatives, which are closely related to the IR and Raman intensities. Thank you very much!
Zhitao
Hi Zhitao,
Yes it can, check the examples and documentation for more information.
Dear Larrucea
I have to thank you for your explanations to this example. I wonder if it is possible to calculate the raman intensities for the raman spectra of a material. In this example we can calculate the raman spectrum but as the frequency of a mode increases, the value on the intensity axis also increases. So how can we know the real intensities for those modes?
Hi Mehmet,
Actually those intensities are wrong. It should be possible to calculate them, but as I recall, it is not so straight forward. Try in the quantum espresso mailing list or something.
Sir, any one can help me where i download quantum expresso windows files and how to install
Sorry for the late reply. There should be some windows binaries on the download page.