Once, I was making some DOS figures with quantum espresso by using the sumpdos.x program by Andrea Ferretti (included in the QE package), but I felt that I am missing some features. Thereby, I wrote my own script, which does something similar, but it is much more automatized, it is faster to use, and it even produces ready-to-publish graphs automatically.
It is part of the quantum espresso distribution, so if you have the source code of quantum espresso, you have it in the …/*espresso*/PP/tools/sum_states.py. If you don’t have it, or you just want to read the source-code online, you can check for sum_states.py in the quantum espresso repository.
So, if you want to plot the DOS with quantum espresso, here are the steps:
- Calculate the wavefunction (single point, with a small “conv_thr”, 10-8 or so)
- Run projwfc.x with an input like:
&projwfc
outdir='.'
prefix='PREF'
ngauss=0, degauss=0.02
DeltaE=0.01
filpdos='sim.proj'
/ - Run the sum_states.py script with something like:
sum_states.py -o out -s "*Fe1*d*" -xr -8.5 5.2
I have run these script with the example of the wustite, and the result is the following one:
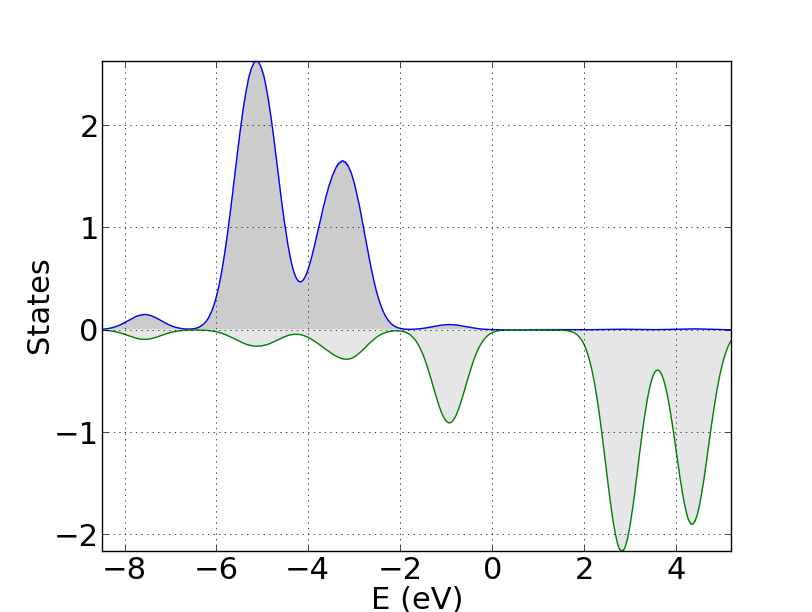
DOS plot produced by sum_states.py
Some other interesting features:
- Tries to find the output files by itself
- Finds out whether there is gnuplot or mathplotlib available, if not, dumps raw data so that you can plot it however you want
- Plots the Up and Down spin in opposite directions and slightly different colors
- Adjusts the range of the ordinate axis for the plot
- Finds out wetter there is X11 forwarding (in case it is executed non-locally) and if there is not, it will try use gnuplot “set terminal dumb”, so that we get an idea of the shape of the data. (… yes, this option is very geek 😛 )
Dear Julen, first of all i want to congratulate with you for your help which you provide to all;
i’m Master degree student of Bionanomaterials at Università Cà Foscari of Venice, and i’m working with QE. What i would like to ask you, about this script, what is the meaning of code written in point 3… i’m using Ubuntu with ifortran for working with QE, might it have problem with the script?
Thanks in advance,
Tommaso.
p.s. i’m not able to use the script, now i list you the steps i had provided:
1)-scf calculation in tetrahedra mode;
2)-dos.x calculation;
3)-projwfc
4)-run your script—> does the pw.out files have to be in the same directory? shall i have to run your script with both the pw.out files and projwfc.out files?
Thanks in advance
Tommaso
Hi Tommaso,
the sum_states script simply plots the electronic density of states for spin-up and spin-down and it can sum the values over the k-space (this is why I originally called it sum_states instead of plot_dos or something like that).
The density of states or DOS tells you about how many electronic states (orbitals) are there at each energy value, so that you can compare values between similar molecules and see how the changes affect on them. Usually, the most useful part is the one located around 0 eV, also known as Fermi level. If you study a semi conductor or non-metallic system, there should be a little interval where the values of both up and down spins is equals to 0, and the horizontal distance between them tells you the HOMO-LUMO gap you can compare with experimental results.
About the second question, first of all, you don’t need to run dos.x, the projwfc.x already provides the local density of states (of each atom and each orbital).
It should work if you use something like this:
sum_states.py -o pw.out -s "*A*B*" -xr -8.5 5.2
where A is the atomic specie you want to check and B the orbital (for example “*Fe*d*).
You can try to grab the list of atoms and orbitals you want by checking with “ls”. For example:
ls *H*s* *O*p*
separating the species with spaces.
I hope it helps.
Thanks a lot!!!
Tommaso
Dear Julen,
I do scf and pdos then I run the python. I have this error and I don’t know why!
#### sum_states.py version 0.2 ####
pylab imported
WARNING: No Fermi energy found. Using 0 e.V. instead
dosfiles list: pdos.proj.pdos_atm#18(H)_wfc#1(s) pdos.proj.pdos_atm#10(H)_wfc#1(s) KAlH4.scf.in pdos.proj.pdos_atm#20(H)_wfc#1(s) pdos.proj.pdos_atm#13(H)_wfc#1(s) pdos.proj.pdos_atm#15(H)_wfc#1(s) pdos.proj.pdos_atm#9(H)_wfc#1(s) pdos.proj.pdos_atm#12(H)_wfc#1(s) pdos.proj.pdos_atm#14(H)_wfc#1(s) pdos.proj.pdos_atm#11(H)_wfc#1(s) pdos.proj.pdos_atm#24(H)_wfc#1(s) pdos.proj.pdos_atm#21(H)_wfc#1(s) pdos.proj.pdos_atm#22(H)_wfc#1(s) pdos.proj.pdos_atm#16(H)_wfc#1(s) pdos.proj.pdos_atm#19(H)_wfc#1(s) pdos.proj.pdos_atm#23(H)_wfc#1(s) pdos.proj.pdos_atm#17(H)_wfc#1(s)
no ksolved
Traceback (most recent call last):
File “sum_states.py”, line 182, in
mati.append([float(line.split()[0]),float(line.split()[1]),float(line.split()[2])])
ValueError: could not convert string to float: calculation
The odd thing is the first time I ran this python every thing was OK and the K-s was plotted. then I tried to use “*” to plot all of them and I had error. Would you please help me? ( I have a K point mesh of 6 8 6. Does it matter?)
Thanks in advance
Samira
Dear Samira,
Check out:
You are giving the PW input file as a DOS file. The script expects the format of all the DOS files to be… well, the one of the DOS files, and that is why it fails.
You should be careful with the wildcards. You could try:
ls YOUR_PATTERN*to check which files are you going to match.
Besides, in case our PW SCF output file is not named “out”, it would be better if you would specify the name of it with the “-o” option. This way it would shift your energy values to the proper ones and you would have the HOMO-LUMO gap at Fermi level (0 eV).
Best regards
Julen
Oh Thanks a lot.
I am such a blind! why I didn’t see it!
I had another mistake too, I should specify which H or K. for example “*1*H*s” because I have 16 H in this file. but I still have this warning:
WARNING: No Fermi energy found. Using 0 e.V. instead
Why?!!specify the name of it with the “-o” and even changed the name of scf output to “out” but it doesn’t work out
(python sum_states.py -o out -s “*5*K*p*” -xr -8.5 5.2)
“*” doesn’t work too. How can I plot all of them together? Can I get any usefull information of such plot?
I am not familiar with python language and I just started QE . sorry for asking so many questions and Thank you again.
I usually name all the PW output files as “out”. I think you have used some other filename (like KAlH4.scf.out).
With the -o option, you enable a little loop which will go trough the output file and check for the line saying “the Fermi energy is…”. Then it parses that value and deducts it from your Kohn-Sham Energies (the ones at the bottom of the Graph) shifting the whole graph to the left.
Oh and my structure is Insulator so just has highest occupation number in output file!
Hahahaha
Thanks so much for taking the time. I really appreciate it
Samira Adimi
Hi,
I am done with first two steps . But I did not got how to incarporate third step ‘sum_states.py -o pw.out -s “*A*B*” -xr -8.5 5.2’ . Can you suggest me in detail how do i use this script. I am working on Linux.
Thanks,
Manjusha
Hi Manjusha,
The use of this script is pretty straight forward.
I assume you have python installed and you are using a terminal with BASH or something similar.
I will try to guess what might have gone wrong.
Make sure your script is executable by :
… and if you haven’t added the directory containing the file to the $PATH (which you probably haven’t), use the whole path to the file. If the script is located in the same folder as your quantum espresso input/output files, add a dot and a slash (“./”) at the beginning of the file name, so that the shell knows where the file to be executed is. So:
Make sure your output is called “pw.out” and that the calculations ended properly. So, they didn’t just exit because they reached the maximum number of steps instead of reaching the convergence criteria.
hi everyone
i am doing cpmd .i dont know how to plot time vs energy at particular temperature.
please help for this case thanks in advance.
Hi Rajkamal,
This is not really related to this post, but anyway…
Take a look to the ENERGIES file. You can use gnuplot and do something like “plot ‘ENERGIES’ using 1:3 w l”